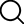3/26/2026
As vital participants in life processes, peptides have always been a research hotspot in the field of chemical synthesis due to the need for efficient and precise synthesis methods. Traditional peptide synthesis methods typically involve the activation of the carboxyl group, converting it into an active ester or other reactive substance, which then couples with an amino group in another molecule. Although this method is widely used, it is usually limited to C→N synthesis and is prone to side reactions such as racemization when involving sensitive amino acids like cysteine. To address this bottleneck, Campagne, J.-M.'s team has taken a different approach, developing a novel peptide synthesis method based on a "reverse activation" strategy—that is, activating the amino group.
As vital participants in life processes, peptides have always been a research hotspot in the field of chemical synthesis due to the need for efficient and precise synthesis methods. Traditional peptide synthesis methods typically involve the activation of the carboxyl group, converting it into an active ester or other reactive substance, which then couples with an amino group in another molecule. Although this method is widely used, it is usually limited to C→N synthesis and is prone to side reactions such as racemization when involving sensitive amino acids like cysteine. To address this bottleneck, Campagne, J.-M.'s team has taken a different approach, developing a novel peptide synthesis method based on a "reverse activation" strategy—that is, activating the amino group.
I. Reverse Thinking from "Activated Carboxylic Acids" to "Activated Amino Acids"
The core of traditional methods lies in activating the carboxyl group, while Campagne, J.-M.'s team cleverly transferred the activation site to the amino group. They used inexpensive and readily available N,N'-carbonyldiimidazole (CDI) in the presence of triethylamine to react with α -amino acid esters, generating and isolating a stable N-imidazolium carboxyl amino acid ester intermediate (Figure 1). This intermediate is equivalent to an "activated amino group" and can be stably stored for several months.
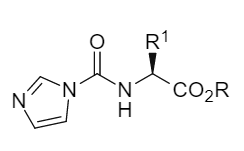
Figure 1 General formula of N-imidazolyl amino acid ester
In subsequent peptide synthesis, this intermediate reacts directly with N-protected amino acids in dichloromethane under mild, alkali-free conditions and in the presence of additives (HOBt/CuBr 2 ), yielding dipeptides in 52-90% yield. This method is compatible with commonly used carbamate protecting groups such as Fmoc, Boc, and Cbz, and racemization was not observed when the reaction substrate involved easily racemic cysteine. However, studies indicate that the conventional peptide synthesis solvent DMF is not suitable for this system.
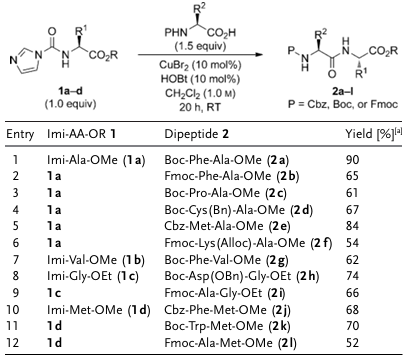
Figure 2. Synthesis of dipeptides via "activated amino groups”
To further validate the potential of this synthetic strategy, the team attempted to synthesize a tetrapeptide via the N→C direction, ultimately obtaining the target product in a 25% yield without finding any racemic products. This discovery demonstrates the significant potential of this method for reverse peptide synthesis, providing a reliable new route for synthesizing complex peptide drugs and bioactive molecules.
II. One-pot synthesis: high efficiency, atom economy, and good stereoselectivity.
Building on previous work, Campagne, J.-M.'s team further optimized the synthetic strategy and developed a one-pot method for synthesizing dipeptides. This method, also carried out under mild, neutral conditions, requires no additional alkali, and generates N-imidazolium carboxyl amino acid ester intermediates in situ. Without the need for separation and purification, these intermediates can react directly with commercially available protected amino acids, significantly simplifying the operational process.
Experimental results show that the one-pot method can also efficiently synthesize a variety of dipeptide derivatives, with yields reaching up to 87%, and is compatible with various N-carbamate protecting groups, exhibiting excellent stereochemical stability. Furthermore, the study found that proline, due to its secondary amine structure, is difficult to synthesize dipeptides through amino activation.
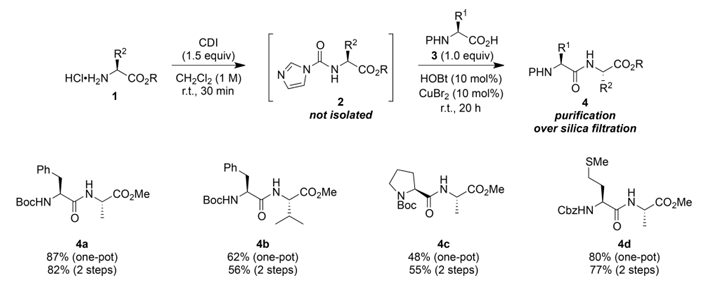
Figure 3 One-pot synthesis of dipeptides
Unlike the two-step method, the one-pot strategy uses the imidazole generated in the system as a base during the "activation of the amino group," forming an imidazole salt, thus eliminating the need for additional base. The study also found that adding an extra base during the coupling process actually hinders the amide bonding reaction between the activated amino group and the carboxylic acid.
III. Microwave-assisted reaction: accelerates reaction and shortens cycle time
To improve synthesis efficiency, Campagne, J.-M.'s team further introduced microwave-assisted technology to explore its application in the "amino activation" strategy.
Based on the two-step synthesis, microwave irradiation was used to promote the bonding reaction of amides (Figure 4). The results showed that microwaves could significantly accelerate the reaction rate, shortening the reaction time from 20 h in the conventional method to 1 h, while maintaining a good yield and without the observation of racemization. Furthermore, the application of this method was broadened to the synthesis of conventional amides, and the yield met expectations.
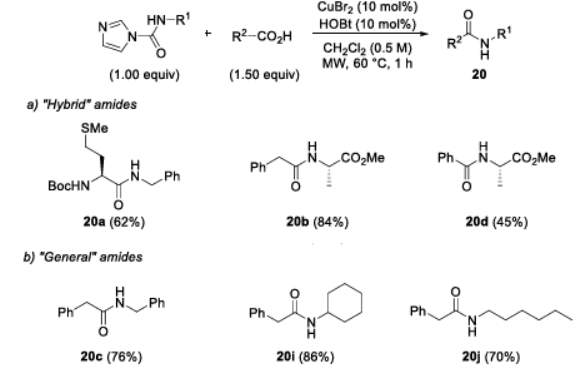
Figure 4 Microwave-assisted synthesis of peptides
Based on experimental observations, the team proposed a possible reasonable reaction pathway (Figure 5): a mixed carbamic anhydride intermediate is formed during the reaction , followed by intramolecular rearrangement and release of carbon dioxide, ultimately generating the target peptide bond.
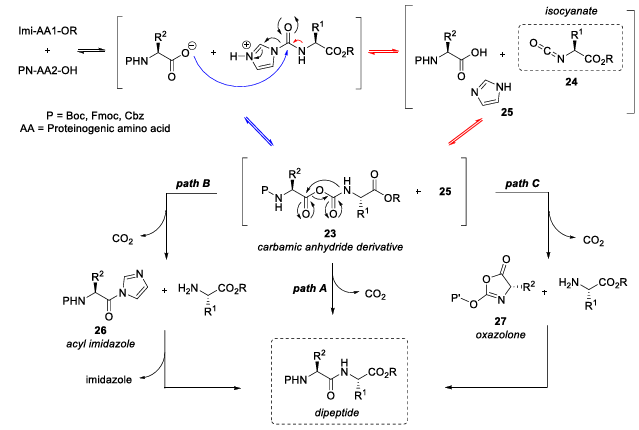
Figure 5 Reaction mechanism
In summary, the CDI-mediated α -amino acid ester activation strategy developed by Campagne, J.-M.'s team provides a mild, practical, and atom-economical new option for peptide bond formation. This method not only effectively avoids the racemization problem of sensitive amino acids during the reaction process but also successfully achieves the challenging N→C direction peptide synthesis. With the continued rise in interest in peptide drug development, this innovative synthetic strategy is expected to become an important addition to researchers' toolboxes.
Company Introduction:
Suzhou Haofan Biotech Co., Ltd. (Stock Code: 301393.SZ), founded in 2003 and headquartered in Suzhou High-tech Zone, is a national high-tech enterprise providing specialty raw materials to pharmaceutical R&D and manufacturing companies worldwide. Its products are mainly used in the synthesis of peptides, nucleotides, and pharmaceuticals, covering a wide range of products including condensing agents for specialty amide bonds, protective agents, linking agents, protein cross-linking agents for antibody-drug conjugates, molecular building blocks, liposomes, and phosphorus reagents. To date, it has cumulatively developed and produced over 1,500 different products.
After more than two decades of unremitting efforts and accumulation, Haofan Biotech has continuously cultivated its expertise in the global peptide synthesis reagent field. It has now developed into a leading enterprise with extensive customized product coverage and significant advantages in large-scale production, capable of meeting the specific needs of various customers. We sincerely invite customers interested in this product to contact us to learn more about product details and explore cooperation opportunities .
References:
[1] Inverse Peptide Synthesis via Activated a-Aminoesters.
DOI: 10.1002/anie.201402147
[2] SequentialOne-PotSynthesisofDipeptidesthroughtheTransient FormationofCDI-N-Protectedα-Aminoesters.
DOI: 10.1002/adsc.201700034
[3] Amine Activation: "Inverse” Dipeptide Synthesis and Amide Function Formation through Activated Amino Compounds.
DOI: 10.1021/acs.joc.2c01288
Please fill out the form below and our sales team will be happy to assist you with a quote on peptide synthesis reagents.